Mechanistic insights into noncanonical protein splicing by a radical SAM splicease
A recent Angewandte Chemie paper by the Vagstad and Piel groups (IMB) illuminates key mechanistic features of the unusual conversion of tyrosine residues to α-keto-β-amino acids by post-translational spliceases. Such ketoamide moieties are clinically used protease-inhibiting pharmacophores.
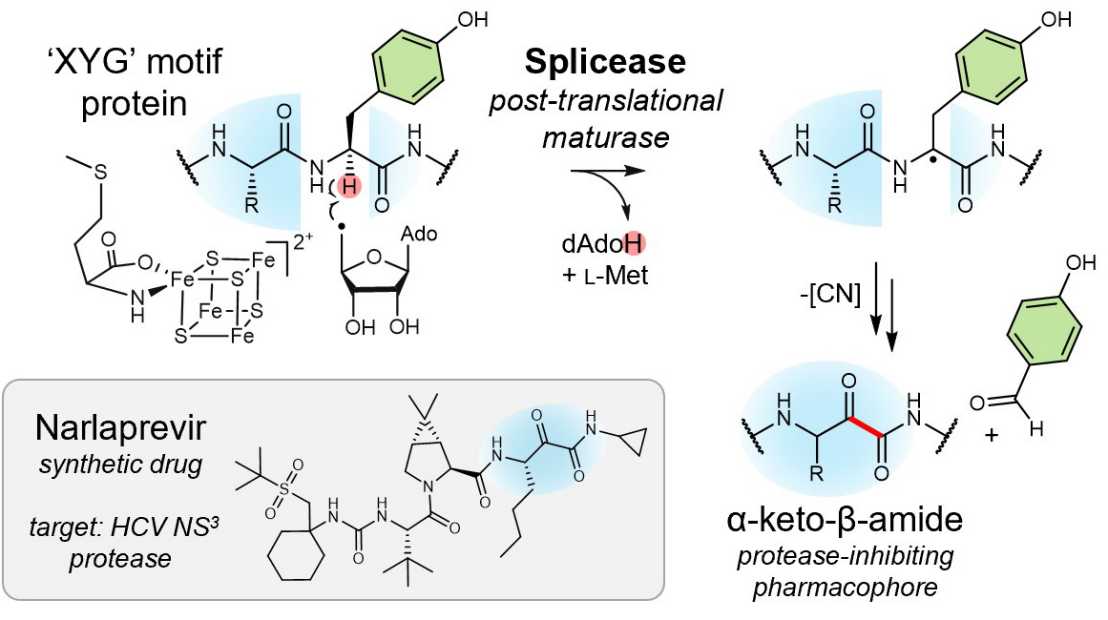
Genome mining of ribosomally synthesized and post-translationally modified peptide (RiPP) natural product pathways has revealed a spectacular array of previously unknown post-translational modifications. An intriguing example is the installation of α-keto-β-amino acids at ‘XYG’ motifs within precursor protein substrates by so-called ‘splicease’ enzymes belonging to the radical S-adenosyl methionine superfamily. Spliceases are the key maturases in a widespread family of RiPP natural products termed spliceotides that were discovered several years ago by the Piel group and comprise potent protease inhibitor activities mediated by the installed ketoamide. The conversion occurs via a formal excision of a tyrosine-derived tyramine unit and reconnection of the peptide backbone—an exceptional reaction with no obvious precedent.
To interrogate the catalytic mechanism, Anna Vagstad and collaborators established the anaerobic in vitro activity for a model cyanobacterial splicease. Using a multidisciplinary approach, the authors found that tyrosine Cα is the initial site of peptide radical formation, 4-hydroxybenzaldehyde is released as the tyrosine-derived coproduct, and that radical intermediates are likely coordinated to an enzyme bound [4Fe-4S] cluster cofactor. These data informed a mechanistic proposal for the biotransformation to diverse α-keto-β-amino acid residues—pharmocophores also present in clinically used peptidomimetic therapeutics, such as the anti-hepatitis C drug narlaprevir.
Link to the paper in external page Angewandte Chemie International Edition.