DIP-MS, a deep interaction proteomics method for the analysis of protein complexes
Deep Interactome Profiling (DIP-MS) offers a sensitive and comprehensive method to study protein complex isoforms concurrently present in affinity purifications. The method combines protein fractionation profiling with machine learning to study modular protein organisation at a high level of detail. The implemented method was developed in the Gstaiger and Aebersold lab (all IMSB) and has been recently published in “Nature Methods”.
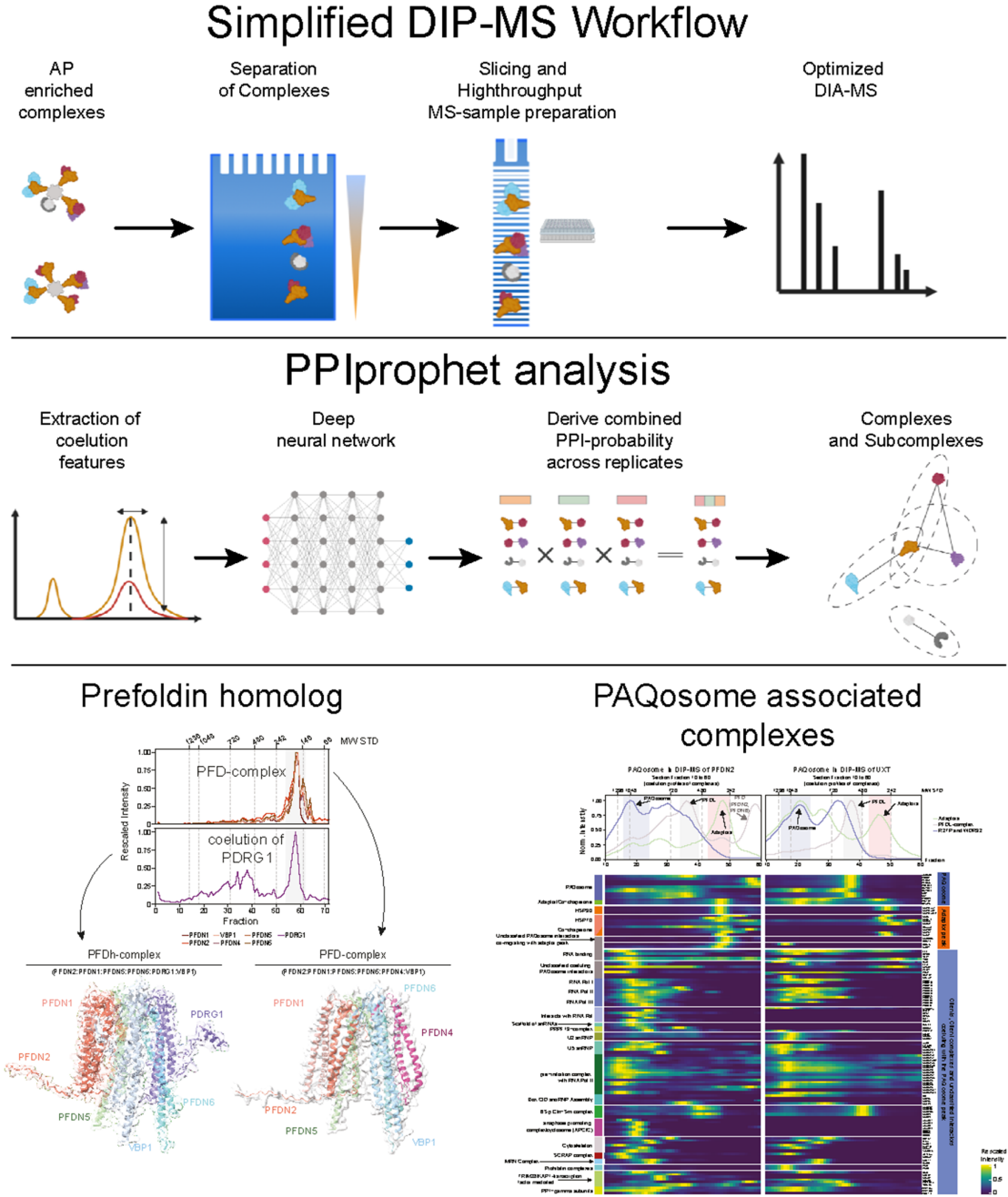
Most proteins are organized in macromolecular assemblies, which represent key functional units regulating and catalysing cellular processes. Despite analytical progress in the systematic collection of binary protein interaction data using affinity purification and mass spectrometry (AP-MS), it is impossible to deconvolute protein complexes from binary protein-protein interaction information obtained from a single AP-MS experiment. To address this problem, the Gstaiger group collected protein coelution profiles from size fractionated affinity purified samples using a high-throughput quantitative mass spectrometry approach. The obtained coelution profiles were analysed by PPIprophet, a deep-learning based framework which was trained on millions of coeluting proteins, in order to identify complex subunits sharing the same co-elution profile.
The new method was tested on the Prefoldin protein family, which constitute important chaperone complexes for the folding of proteins and protein complexes. DIP-MS, besides identification of all Prefoldin related complexes known from the literature, enabled the identification of a novel Prefoldin related complex isoform and the characterization of multiple sub-assemblies as well as client proteins from the coelution data obtained from a single affinity purification experiment. The presented results demonstrate that DIP-MS will have great potential to reveal complex formation at unprecedented depth and resolution for a broad range of proteins.
Link to paper in external page "Nature Methods".